-
学习资料来源:
-
官网:https://scanpy-tutorials.readthedocs.io/en/latest/spatial/basic-analysis.html【注意教程有两个版本,这里是latest版本的学习笔记】
这篇教程主要介绍怎么使用scanppy进行空间转录组数据分析,以10X数据为主,顺带介绍MERFISH数据的分析。
数据读取
本次分析使用的数据为人类淋巴结,数据下载地址:https://support.10xgenomics.com/spatial-gene-expression/datasets/1.0.0/V1_Human_Lymph_Node
函数datasets.visium_sge()可以下载10x官网的数据,如果是你自己的数据,可以使用sc.read_visium()读取进来,返回一个AnnData对象。
命令版本:
import scanpy as sc
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
sc.logging.print_versions()
sc.settings.verbosity = 3
outdir = '/Pub/Users/project/scanpy/stRNA/'
adata = sc.datasets.visium_sge(sample_id="V1_Human_Lymph_Node")
adata.var_names_make_unique()
adata.var["mt"] = adata.var_names.str.startswith("MT-")
sc.pp.calculate_qc_metrics(adata, qc_vars=["mt"], inplace=True)
# Visium data数据结构
adata
数据情况如下:
AnnData object with n_obs × n_vars = 4035 × 36601
obs: 'in_tissue', 'array_row', 'array_col', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_50_genes', 'pct_counts_in_top_100_genes', 'pct_counts_in_top_200_genes', 'pct_counts_in_top_500_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt'
var: 'gene_ids', 'feature_types', 'genome', 'mt', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts'
uns: 'spatial'
obsm: 'spatial'
QC以及预处理
基于总的counts与基因,对spots进行一些基本的过滤:
fig, axs = plt.subplots(1, 4, figsize=(15, 4))
sns.distplot(adata.obs["total_counts"], kde=False, ax=axs[0])
sns.distplot(adata.obs["total_counts"][adata.obs["total_counts"] < 10000], kde=False, bins=40, ax=axs[1])
sns.distplot(adata.obs["n_genes_by_counts"], kde=False, bins=60, ax=axs[2])
sns.distplot(adata.obs["n_genes_by_counts"][adata.obs["n_genes_by_counts"] < 4000], kde=False, bins=60, ax=axs[3])
plt.savefig(outdir + "01-sns.distplot.png")
结果:
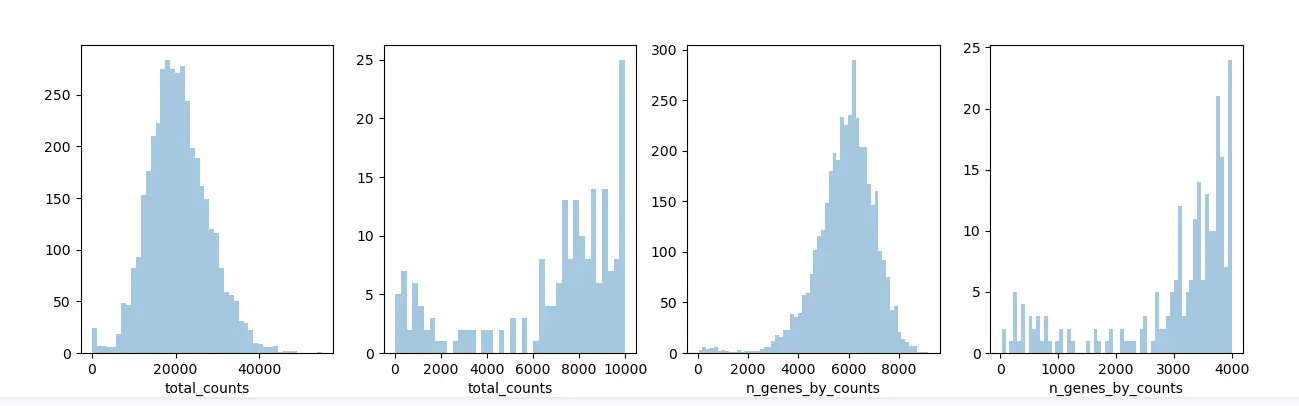
过滤:
-
counts:大于5000,小于35000
-
min_cell:10
-
mt:20%
sc.pp.filter_cells(adata, min_counts=5000)
sc.pp.filter_cells(adata, max_counts=35000)
adata = adata[adata.obs["pct_counts_mt"] < 20]
print(f"#cells after MT filter: {adata.n_obs}")
sc.pp.filter_genes(adata, min_cells=10)
使用normalize_total对Visium Counts数据进行标准化,然后进行高变基因选择。此外,对于数据标准化还可以选择:SCTransform or GLM-PCA。
sc.pp.normalize_total(adata, inplace=True)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata, flavor="seurat", n_top_genes=2000)
基于相似性对数据进行降维聚类
聚类:
sc.pp.pca(adata)
sc.pp.neighbors(adata)
sc.tl.umap(adata)
sc.tl.leiden(adata, key_added="clusters")
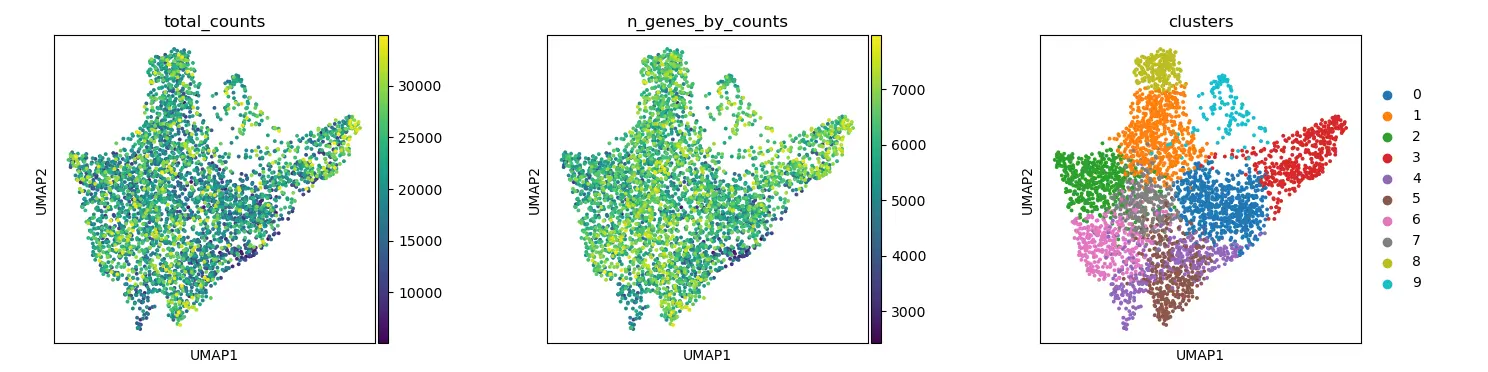
我们绘制一些协变量来检查 UMAP 中是否有任何与总Counts和检测到的基因相关的特定结构。
plt.rcParams["figure.figsize"] = (4, 4)
sc.pl.umap(adata, color=["total_counts", "n_genes_by_counts", "clusters"], wspace=0.4)
plt.savefig(outdir + "02-umap.png")
结果图:
空间坐标可视化
看看 total _ count 和 n _ gene _ by _ count 在空间坐标系中是如何表现的,可以使用 sc.pl.space 函数覆盖所提供的H&E染色图像顶部的圆形Spots
plt.rcParams["figure.figsize"] = (8, 8)
sc.pl.spatial(adata, img_key="hires", color=["total_counts", "n_genes_by_counts"])
plt.savefig(outdir + "03-spatial.png")
结果图:

sc.pl.spatial函数附加参数:
-
img_key:adata.uns对象中的值
-
crop_coord:用于裁剪的坐标(左、右、上、下)
-
alpha_img:图像透明度的 alpha 值
-
bw:将图像转换为灰度
-
size:spot的大小
通过在空间维度上可视化聚集样本,我们可以深入了解组织的组织结构,并有可能进入细胞间通讯
sc.pl.spatial(adata, img_key="hires", color="clusters", size=1.5)
plt.savefig(outdir + "03-spatial_clusters.png")
结果图:在基因表达空间中属于同一cluster的spots常常在空间维度上同时出现。例如,属于cluster 3的spots通常被属于cluster 0的spots所包围(与官网颜色不一致)
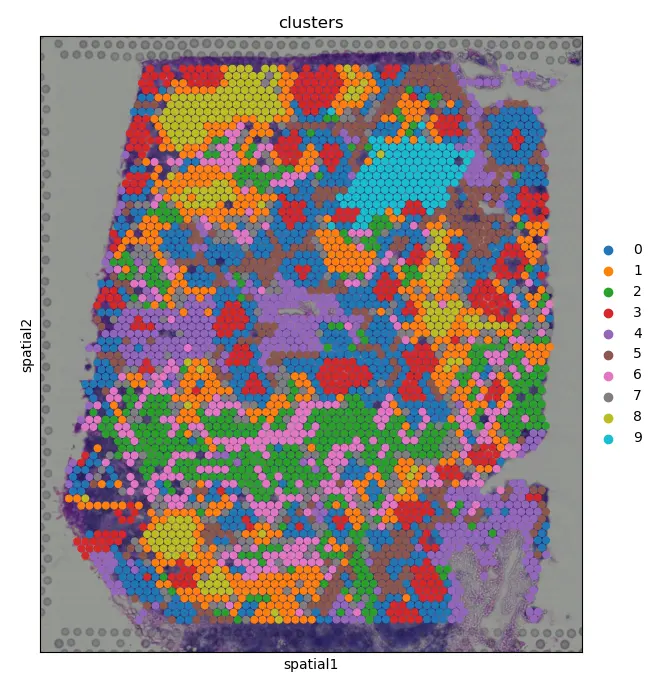
我们可以放大感兴趣的特定区域,以获得定性的见解:
- crop_coord:Coordinates to use for cropping the image (left, right, top, bottom)
sc.pl.spatial(adata, img_key="hires", color="clusters", groups=["0", "3"], crop_coord=[1200, 1700, 1900, 1000], alpha=0.5, size=1.2)
plt.savefig(outdir + "03-spatial_clusters_0_3.png")
Cluster marker genes
让我们进一步观察集群3,它出现在图像中的小群spots中。
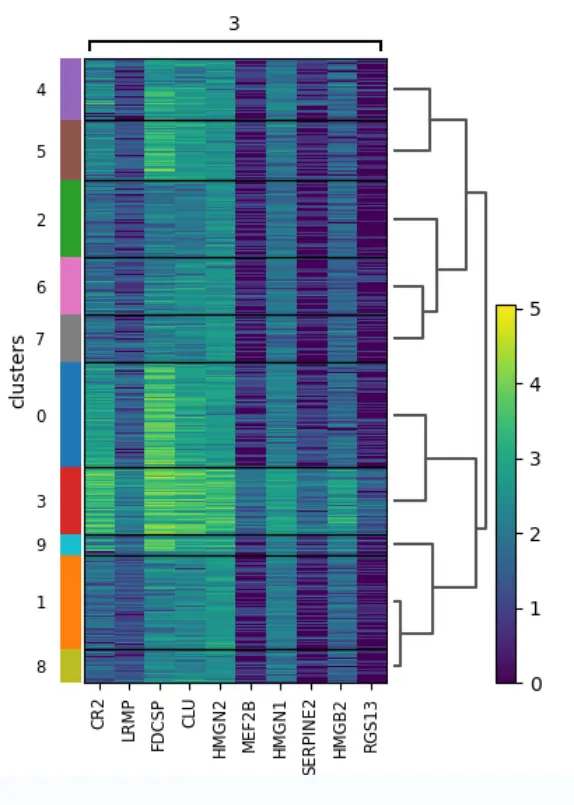
计算标记基因并绘制热图,其中包含前10个标记基因在整个cluster中的表达水平:
sc.tl.rank_genes_groups(adata, "clusters", method="t-test")
sc.pl.rank_genes_groups_heatmap(adata, groups="3", n_genes=10, groupby="clusters")
plt.savefig(outdir + "04-rank_genes_groups_heatmap.png")
结果图:我们看到 CR2概括了空间结构
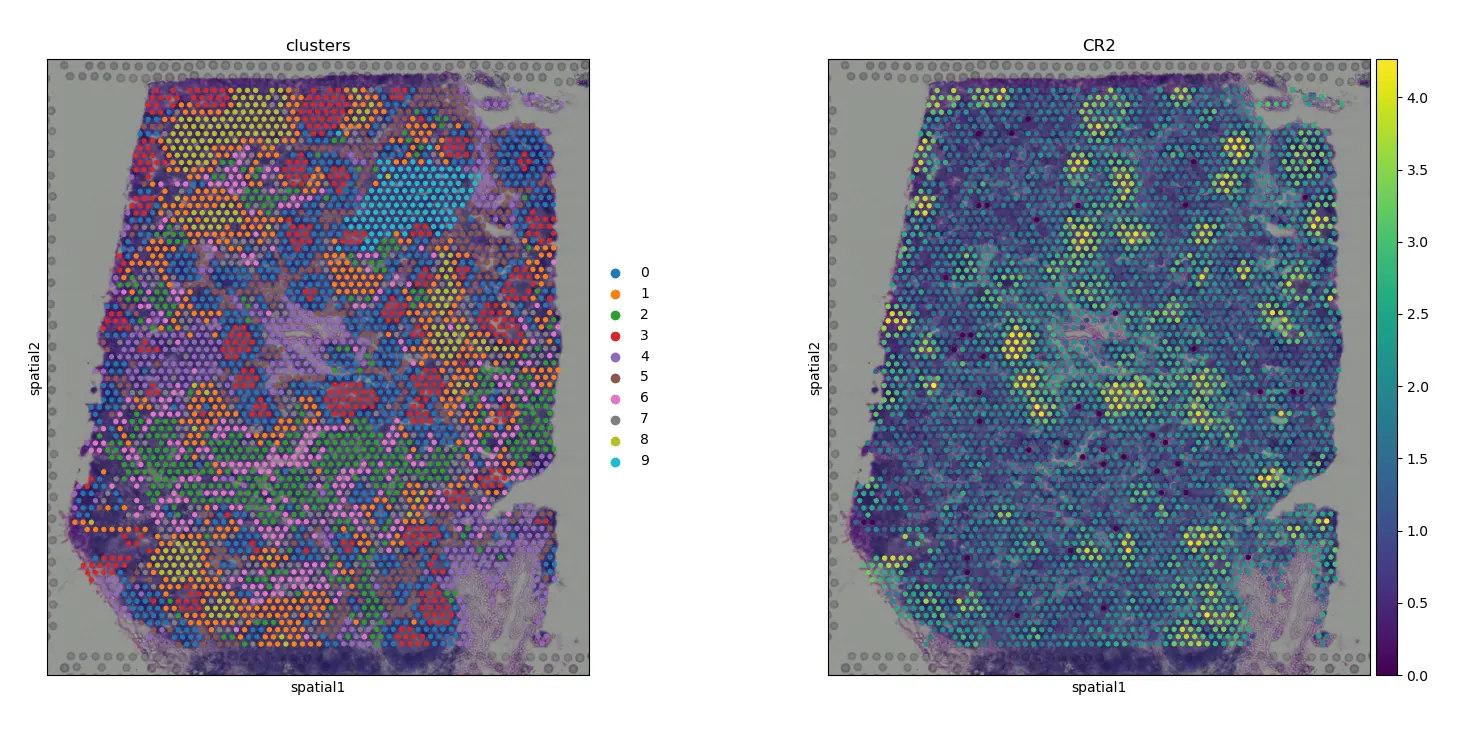
可视化CR2基因表达:
sc.pl.spatial(adata, img_key="hires", color=["clusters", "CR2"])
plt.savefig(outdir + "04-spatial_CR2.png")
结果图:
空间可变基因
空间转录组学允许研究人员调查基因表达趋势如何在空间中变化,从而确定基因表达的空间模式。
为此,我们使用 SpatialDE Svensson18(代码) ,一个基于高斯过程的统计框架,旨在识别空间可变基因。
# 安装在scanpy的conda环境中
pip install spatialde
还有其他几个可识别空间可变基因的工具:
首先,将归一化的counts和坐标转换为pandas dataframe,这是spatialDE所需的输入
运行 SpatialDE 需要相当长的时间:
import SpatialDE
counts = pd.DataFrame(adata.X.todense(), columns=adata.var_names, index=adata.obs_names)
coord = pd.DataFrame(adata.obsm['spatial'], columns=['x_coord', 'y_coord'], index=adata.obs_names)
results = SpatialDE.run(coord, counts)
将结果与变量注释的 DataFrame 连接起来
results.index = results["g"]
adata.var = pd.concat([adata.var, results.loc[adata.var.index.values, :]], axis=1)
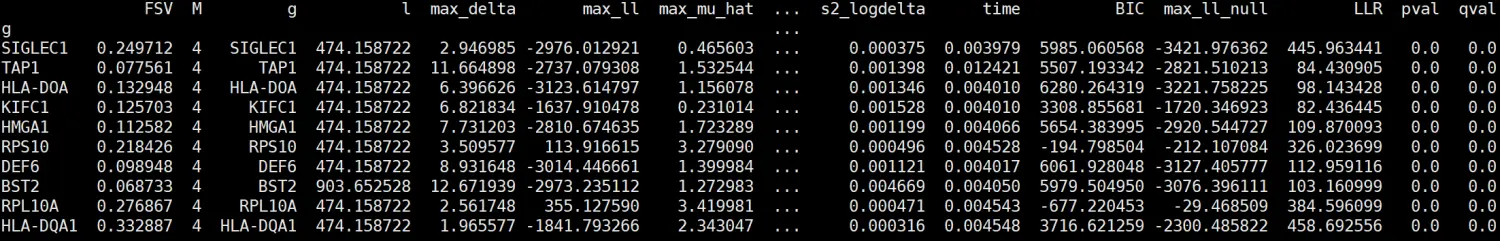
检查在空间上变化的重要基因,并用 sc.pl.space 函数将它们可视化
results.sort_values("qval").head(10)
结果:
选择两个可视化:
sc.pl.spatial(adata, img_key="hires", color=["KIFC1", "TAP1"], alpha=0.7)
plt.savefig(outdir + "05-spatial_FSV.png")
MERFISH数据示例
如果使用基于 FISH 技术生成的空间数据,只需读取坐标并将其分配给 adata.obm 元素
来看看这个数据的例子:Xia et al. 2019
首先从原文下载坐标和counts数据
import urllib.request
url_coord = "https://www.pnas.org/highwire/filestream/887973/field_highwire_adjunct_files/15/pnas.1912459116.sd15.xlsx"
filename_coord = "pnas.1912459116.sd15.xlsx"
urllib.request.urlretrieve(url_coord, filename_coord)
url_counts = "https://www.pnas.org/highwire/filestream/887973/field_highwire_adjunct_files/12/pnas.1912459116.sd12.csv"
filename_counts = "pnas.1912459116.sd12.csv"
urllib.request.urlretrieve(url_counts, filename_counts)
## 没下载下来,先去手动下下来好了
读取数据
dir = '/Pub/Users/project/scanpy/data/'
coordinates = pd.read_excel(dir+"pnas.1912459116.sd15.xlsx", index_col=0)
counts = sc.read_csv(dir+"pnas.1912459116.sd12.csv").transpose()
adata_merfish = counts[coordinates.index, :]
adata_merfish.obsm["spatial"] = coordinates.to_numpy()
adata_merfish
AnnData object with n_obs × n_vars = 645 × 12903
obsm: 'spatial'
接着进行标准的预处理与降维
sc.pp.normalize_per_cell(adata_merfish, counts_per_cell_after=1e6)
sc.pp.log1p(adata_merfish)
sc.pp.pca(adata_merfish, n_comps=15)
sc.pp.neighbors(adata_merfish)
sc.tl.umap(adata_merfish)
sc.tl.leiden(adata_merfish, key_added="clusters", resolution=0.5)
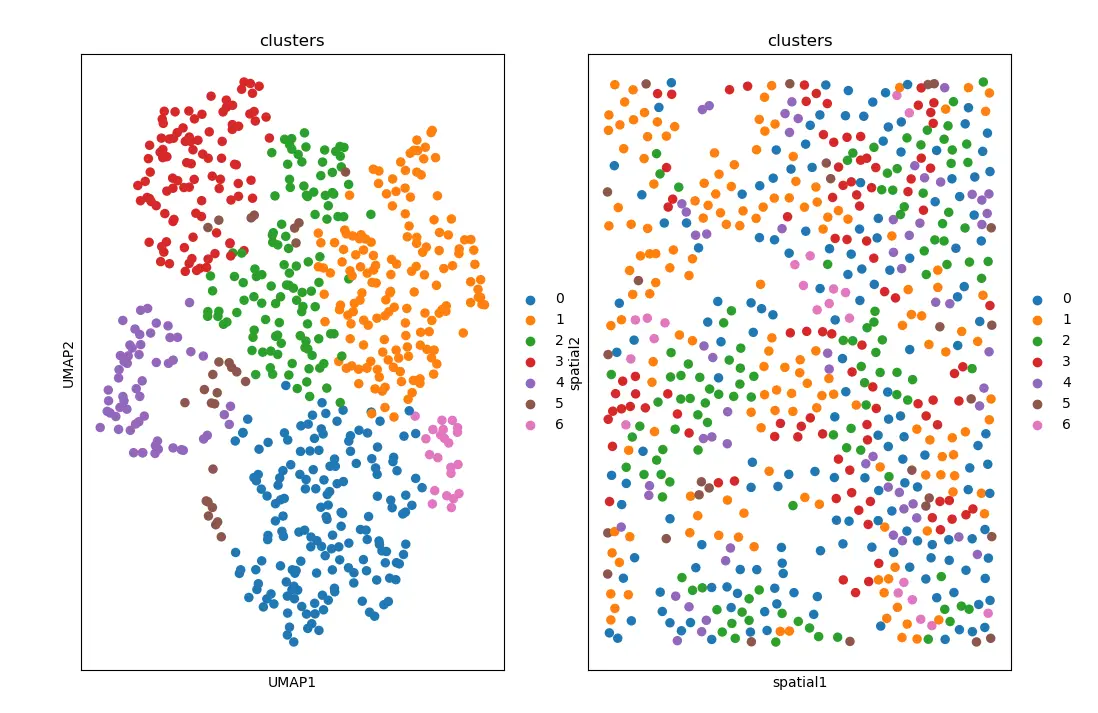
这个单细胞数据来自只有一种细胞类型的cultured U2-OS cells。聚类后的cluster包含细胞周期中不同阶段的细胞states,鉴于实验的设置,我们不期望在空间维度上看到具体的结构。
我们可以在UMAP空间和这样的空间坐标上可视化Leiden得到的cluster。
fig, axs = plt.subplots(1, 2, figsize=(12, 8))
sc.pl.umap(adata_merfish, color="clusters", ax=axs[0])
sc.pl.embedding(adata_merfish, basis="spatial", color="clusters", ax=axs[1])
plt.savefig(outdir + "06-adata_merfish_spatial.png")
结果图:
希望你觉得这个教程很有用!